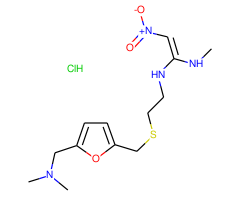
基本信息
| 分子式 | C13H22N4O3S·HCl |
| 分子量 | 350.86 |
| MDL编码 | MFCD00069339 |
| Merck | 14,8110 |
| EINECS 编号 | 266-333-0 |
产品描述
Ranitidine是组胺H2-受体拮抗剂,IC50为3.3±1.4 μM。
靶点(IC50 & Targe)
H2 receptor
体外研究
Ranitidine使肝细胞从激活的中性粒细胞的毒性产物 被杀死,而Famotidine缺乏这种能力。[1] Ranitidine抑制脂多糖体外刺激的单核细胞产生肿瘤坏死因子-α(TNF-α)。[2] 在分离的豚鼠肝细胞中,Ranitidine剂量依赖性地减少吗啡Kel,最大效应为50%,并增加吗啡-6-葡糖苷酸和吗啡-3-葡糖苷酸的相对浓度。Ranitidine逐渐减小吗啡-3-葡糖苷酸/吗啡-6-葡糖苷酸比高达21%。[3]
体内研究
在大鼠中,Ranitidine导致肝损伤,证据是6小时内增加的血清丙氨酸转氨酶,天冬氨酸转氨酶和γ-谷氨酰转移酶活性。[1] 在大鼠中,Ranitidine导致肝损伤,证据是6小时内增加的血清丙氨酸转氨酶,天冬氨酸转氨酶和γ-谷氨酰转移酶活性。[2] 在LPS/ RAN处理的大鼠中,Ranitidine在肝损伤之前增强LPS诱导的凝固,以及抗凝血剂减少肝损伤。在肝血窦的形成过程中,Ranitidine/LPS处理导致大鼠纤维蛋白凝块,并预防与减少的肝细胞损伤相关的纤维蛋白沉积。在大鼠中,Ranitidine在肝细胞损伤前增强LPS诱导的TNF的增加。[4] 在大鼠中,Ranitidine在高架十字迷宫中表现抗焦虑作用,表现在更多的时间留在开放臂上,更多的最终偏移。[5]
参考文献
[1] Luyendyk JP, et al. J Pharmacol Exp Ther,?003, 307(1), 9-16.
[2] Okajima K, et al. J Pharmacol Exp Ther,?002, 301(3), 1157-1165.
[3] Aasmundstad TA, et al. Pharmacol Toxicol, 1998, 82(6), 272-279.
[4] Tukov FF, et al. Toxicol Sci,?007, 100(1), 267-280.
[5] Privou C, et al. Neuropharmacology,?998, 37(8), 1019-1032.
安全信息
| 图形或危害标志 |   |
| 提示语 | Warning |
| 危险说明 | H302 H410 |
| 防范说明 | P264 P270 P273 P301+P312 P391 P501 |
| UN号码 | 3077 |
| 危险分类 | 9 |
| 包装等级 | III |
| WGK | 2 |
| RTECS | KM6557000 |
| TSCA | 否 |
百灵威科技

 m.cnreagent.com
m.cnreagent.com